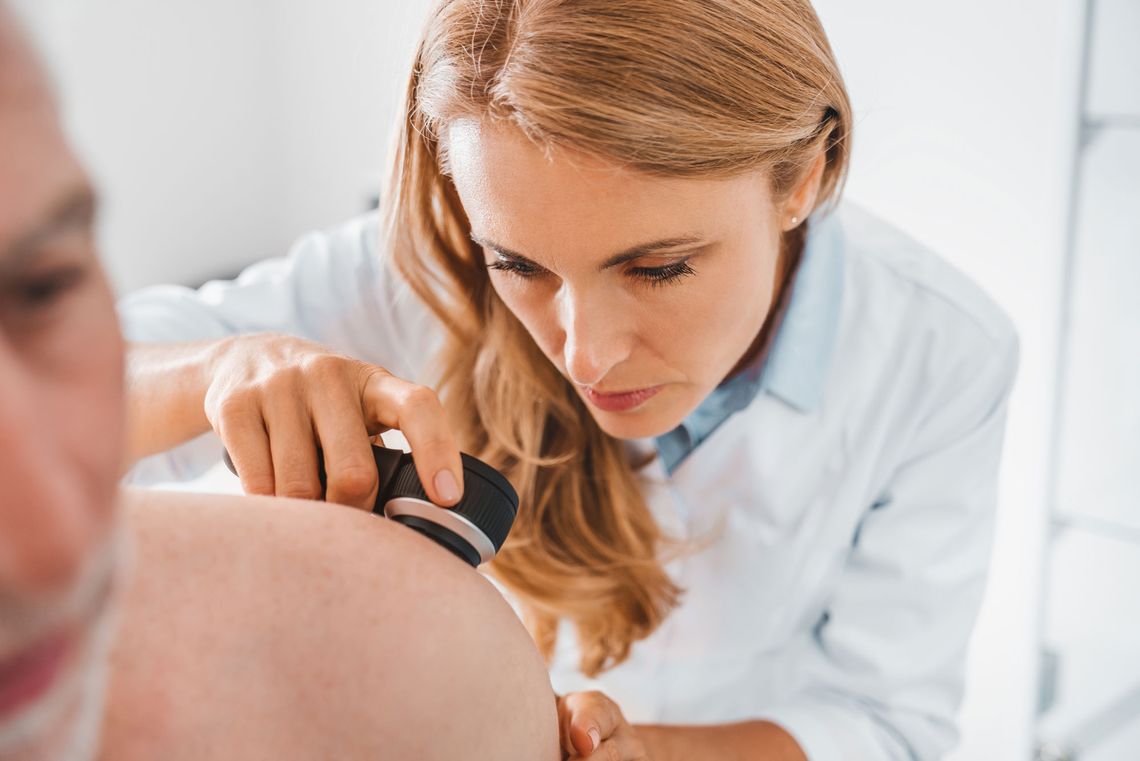
We are SGS proderm - your contract research organization for dermatology. As the 'European Center of Excellence for Dermatology Research' of SGS, the world leader in testing, inspection and certification, we are dedicated to conducting clinical trials. Utilizing our scientific expertise, methodological innovations, and cutting-edge technologies, we establish pioneering benchmarks in product testing.





![[Übersetzen nach: Englisch] Anti Aging Research](/fileadmin/_processed_/5/3/csm_lcoct_collagen_attenuation_ec106b4e9e.jpg)

![[Übersetzen nach: Englisch] Ehemaliges und neues Top-Management](/fileadmin/_processed_/6/2/csm_IMG_2857_web_ac51e56e96.jpg)

![[Übersetzen nach: Englisch] Measurement of well being effects](/fileadmin/_processed_/7/7/csm_eeg_well_being_2a916b7ce6.jpg)

![[Übersetzen nach: Englisch] news logo](/fileadmin/_processed_/8/a/csm_pr_announcement_acquisition_16_9_64ef1d5e2e.jpg)




![[Übersetzen nach: English] proderm lc oct](/fileadmin/_processed_/9/a/csm_lc_oct_listenansicht_e61505ffa4.jpg "[Übersetzen nach: English]")
![[Übersetzen nach: English] proderm new brand design](/fileadmin/_processed_/2/8/csm_new_brand_design_v2_57aae9fc5d.jpg)
![[Übersetzen nach: English] proderm Nachhaltigkeitsbericht](/fileadmin/_processed_/5/a/csm_nachhaltigkeitsbericht21_d056410424.jpg)
![[Übersetzen nach: English] proderm LC-OCT Methode](/fileadmin/_processed_/2/d/csm_line-oct_57f8037e19.jpg)
![[Übersetzen nach: English] proderm Kooperation Newtone](/fileadmin/_processed_/6/6/csm_Newtone-Colorface-shooting-_a03e83df30.jpg)
![[Übersetzen nach: English] proderm site Elmshorn](/fileadmin/_processed_/9/6/csm_elmshorn_karte_300e42d7cc.jpg)


![[Übersetzen nach: English] case study androgenetic alopecia](/fileadmin/_processed_/4/2/csm_androgenetic_alopecia_listenansicht_9806fed0ae.jpg)


![[Übersetzen nach: English] Medizinprodukteverordnung EU](/fileadmin/_processed_/a/a/csm_EU_MDR_669e34136d.jpg "[Übersetzen nach: English] Medizinprodukteverordnung EU")
![[Übersetzen nach: English] Entwicklung von Pflastern](/fileadmin/_processed_/4/b/csm_medizinprodukte_e241baf5a4.jpg)
![[Übersetzen nach: English] proderm Umbau Luna](/fileadmin/_processed_/9/7/csm_Solaris_luna_2_d19392f7e3.jpg "[Übersetzen nach: English] proderm Umbau Luna")
Academy

Webinars
Do they deliver what they promise?
-
Online
This will be a virtual conference. Details about the platform will be provided after completion of the registration.
