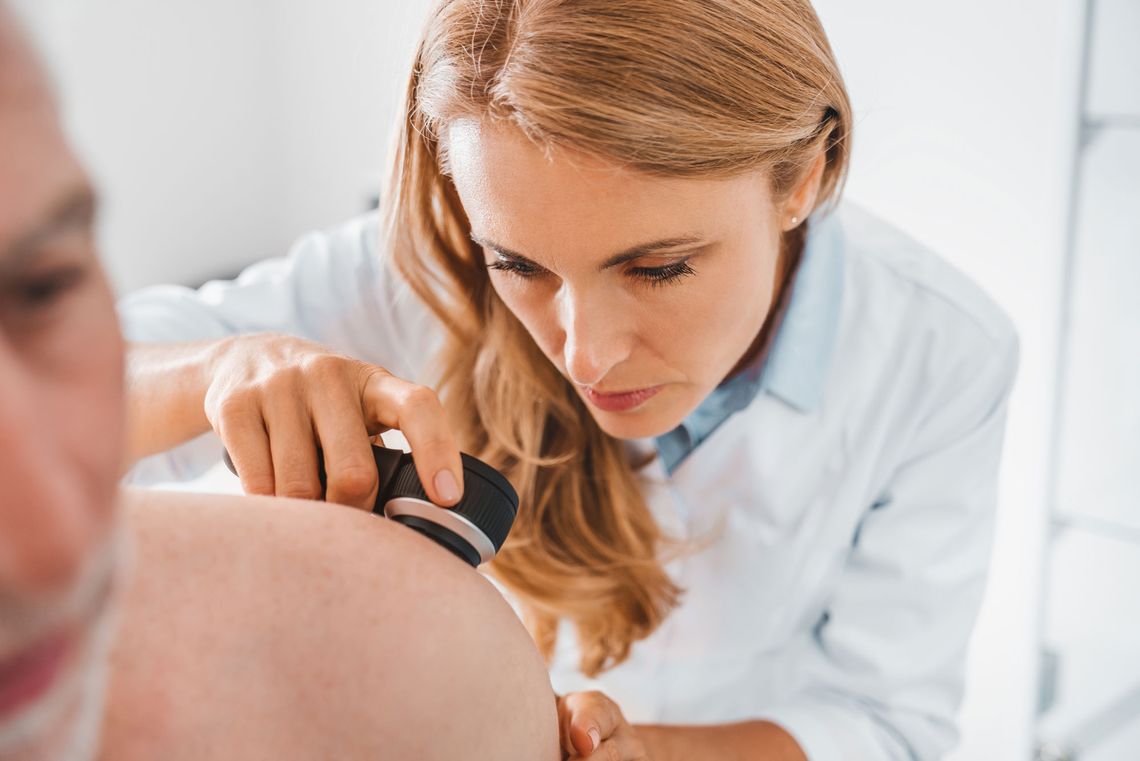
Wir sind SGS proderm – Ihr Auftragsforschungsinstitut für Dermatologie. Als europäisches Exzellenzzentrum der SGS, dem Weltmarktführer für Prüfung, Inspektion und Zertifizierung, widmen wir uns der Durchführung klinischer Studien. Dabei setzen wir mit wissenschaftlicher Expertise, methodischen Innovationen und Spitzentechnologien neue Maßstäbe.


News








Scientific Consulting, Medical, SGS proderm
proderm veröffentlicht Ergebnisse aus klinischen Studien




Medical, SGS proderm
proderm etabliert Kooperation mit dermatologischer Fachpraxis "Prager & Partner"


Scientific Consulting, Consumer Care, Medical, SGS proderm
Einzigartige Bildgebung und maximale Effizienz










Academy

Webinare
Halten sie, was sie versprechen?
-
Online
This will be a virtual conference. Details about the platform will be provided after completion of the registration.
